
Reelin groovy
Chromosome diseases disrupt many genes at once, but some genes matter more than others. 8p iPSC-derived neurons and brain organoids converge on a candidate driver gene located on chromosome 7: Reelin.


In collaboration with


As discussed in the 8p CureMap v1 published last year, the driver gene concept defines the way we think about chromosome diseases.
For example, in trisomy 21 aka Down Syndrome, there is an extra third copy of chromosome 21. Curiously, only some genes on chromosome 21 are dosage-sensitive, e.g., Dyrk1a, meaning the cell stresses out because of the over-expression (or under-expression in the case of chromosomal deletions) of some genes, not others. Put another way, the body doesn’t seem to care if all chromosome 21 genes are in excess, just a select handful of driver genes.
An alternative conceptual framework for dissecting the genetic basis of chromosome diseases is that while there may be driver genes responsible for specific life-altering symptoms or congenital malformations, rather it’s the cumulative burdens of aneuploidy experienced by every cell sum up to 8p disease.
An exacerbating complication is that genes physically located on other chromosomes — ostensibly beyond the blast radius of the damaged 8p chromosome — get hit in the cross-fire either because of transcriptional network entanglement or via long-distance physical interactions between genetic elements on different chromosomes.
Of course, the messy reality for 8p disease probably involves contributions from all three mental models: one or more driver genes on the damaged chromosome; one or more driver genes on unaffected chromosomes; aging-related cell stresses caused by aneuploidy.
As part of the disease modeling segment of the Two-Year Research Plan, Project 8p has funded two complementary approaches using iPSCs from each member — mom, dad, child — of a pioneer 8p family trio. Hereafter the child is referred to as the proband, and parents are referred to as controls.
One set of vials was shipped to a biotech startup in the Bay Area, and another set was sent to a translationally focused academic lab in SoCal. The first approach was performed in Palo Alto by Rarebase PBC, whose platform Function leverages 2-D glutamatergic excitatory cortical neurons (“iNeurons”) cultured for 1-3 weeks in the lab. Here’s quality control data indicating successful differentiation of iPSCs into iNeurons.

The second approach was performed in the lab of Professor Alysson Muotri at UCSD. The Muotri lab has many years of expertise in long-term culturing of 3-D brain organoids derived from patient iPSC lines. As a first of its-kind-experiment, the Muotri lab grew up brain organoids from all three members of a family trio for up to 9 months!

Let’s examine the Rarebase data first. As a sanity check, we first confirmed that the 8p genes in the deleted and duplicated regions behave as expected.
In other words, the mRNA expression of 8p genes located in the deleted region should be ~50% of normal, e.g., CSMD1 (orange box), while the mRNA expression of 8p genes located in the duplicated region, e.g., FGFR1 (blue box), should be ~200% of normal. So far, so good.
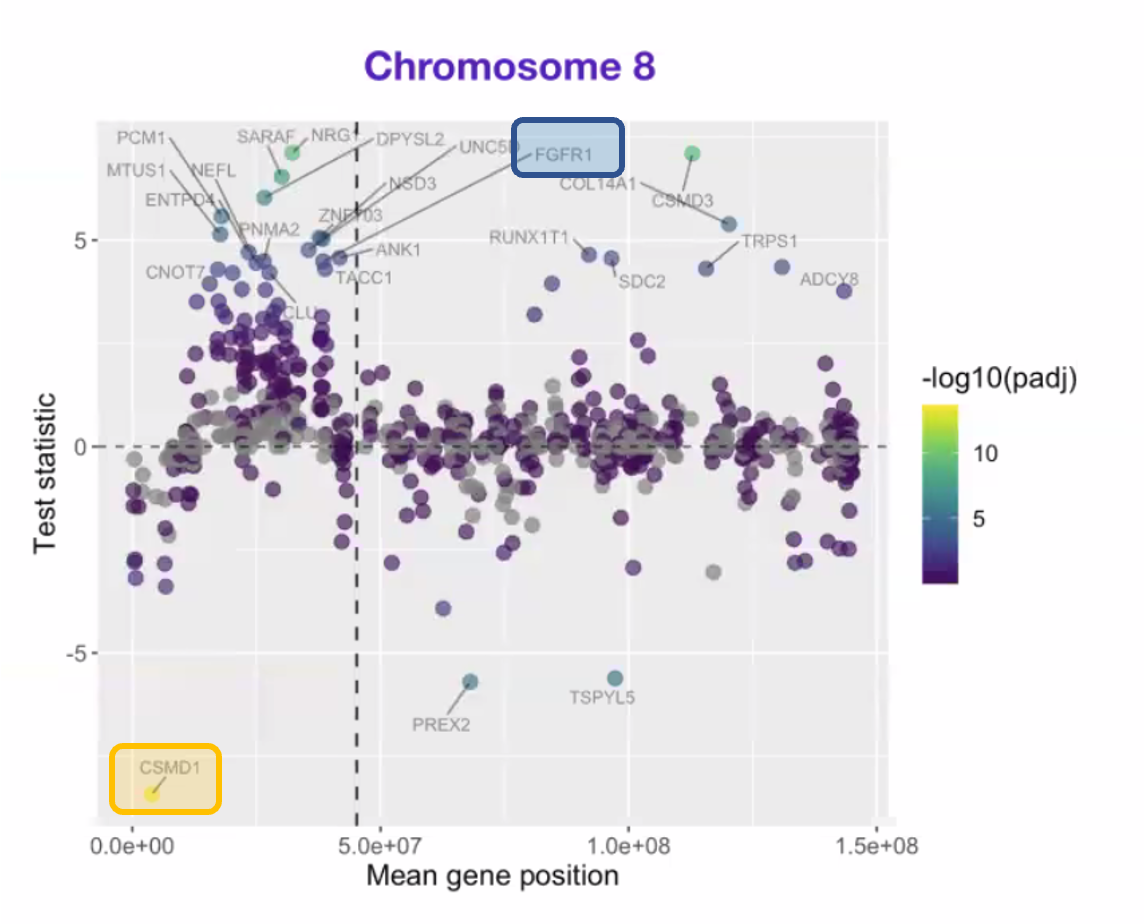
Next, a genome-wide analysis below shows the expression of genes across all 23 pairs of chromosomes. Note the absence of any signal from the Y chromosome since the child/proband is female.
Chromosome 8 is highlighted (second row, center), as is chromosome 7 to the left of chromosome 8, and chromosome 20 (fourth row). We’ll explain in a moment why chromosome 7 is highlighted. Chromosome 20 is more concerning potentially.
There’s a duplication of the long arm of chromosome 20 that recurs in the generation of iPSC lines, as our 8p researcher Professor Stefan Pinter pointed out in last month’s 8p Research Newsletter. This duplication confers a growth advantage during iPSC generation and/or passage in culture, and therefore may be unavoidable in this model system. As a remedy, that’s why we generate independent clones of the same iPSC line. One clone is not enough!

The following plots show bulk RNAseq data from 8p proband iNeurons cultured for one week (Day 7) or three weeks (Day 21) and normalized to the maternal line. Data from the paternal line will be processed soon and will hopefully replicate these results.
Reassuringly, the genes located in chromosome 8p behave as expected. For example, you can see CSMD1 is under-expressed at both Day 7 and Day 21. CSMD1 has been on our radar as an 8p driver gene since the Okur paper (Okur, V. et al 2021).
OPRM1, which encodes the mu opioid receptor, is statistically significantly over-expressed at both Day 7 and Day 21. Two genes are over-expressed only at Day 21: CARTPT and RELN, which is short for Reelin. CARTPT stands for “Cocaine- and amphetamine-regulated transcript.”

Zooming in on the differences between the two timepoints yields this plot. Clearly all three genes — OPRM1, CARTPT and RELN/Reelin — pass the sniff test in terms of genes that may be playing a physiological role in 8p disease.

However, without unbiased confirmation that any of those three genes is dysregulated in a different 8p disease model — ideally a more complex model like brain organoids differentiated from the same pioneer 8p family trio — all we have is a preliminary result.
Sometimes in science, lightning strikes twice. That’s usually the sign of a discovery. In the same week in late January, we learned that Reelin/RELN was the star of the brain organoid dataset.
Here’s the summary slide carefully prepared by Prof Muotri who presented at the January 8p Research Roundtable, the kickoff for the 2023 season. Busier than the New York City subway map, only connoisseurs of UMAP plots will appreciate the finer details.

For our purposes, let’s zoom in on the plot summarizing the cell proportions over time.

At the 1-month and 3-month timepoints, the cell composition of the family trio of organoids are mostly the same. Early data suggested increased apoptosis in the proband, the brain organoid version of microcephaly. But brain organoids of one of the controls (JE2) were smaller than the other control (JE1), so best not to over-interpret on the basis of a single trio.
Going back to the cell composition breakdown at 9 months, there’s a striking population of glutamatergic RELN+ neurons that are not present in mom or dad brain organoids. The late-onset of Reelin expression in both the brain organoids and iNeurons is also noteworthy.
However, like any result from a 9-month brain organoid from a single clone of a single proband’s iPSC line, we need to replicate this result in another 8p family trio. Reelin over-expression must also be confirmed by at the protein level and also by immunohistochemistry. If the Reelin result is real, we’re just at the beginning.
Of course it could be very well be a false positive, or a mischievous experimental artifact. An alternative framing is that we know Reelin meets the definition of a dosage-sensitive gene to a tee. It’s been most studied in the context of its under-expression or loss-of-function.
But what happens when there’s overactive Reelin or too much Reelin at the wrong time, say too much in fetal development? If you get past the neurodevelopmental window and the slide toward neurodegeneration begins, could there actually be a protective advantage to increased Reelin activity?
Data published in 2011 from a Reelin-overexpressing mouse model suggests a protective effect. Data published just yesterday shows that a rare gain-of-function Reelin variant protected a person against early-onset Alzheimer’s.
This is why we’re Reelin groovy.